平台1:中南大学 相图相变及材料智能设计科学中心
平台2:中德“电化学存储系统集成计算材料工程”联合实验室
平台3:中德“微结构”联合实验室
http://imdpm.csu.edu.cn
一、中心介绍
中南大学“相图、相变及材料智能设计科学中心”由杜勇教授于2003年1月回国后组建。已形成一支由4名教授(教育部长江学者特聘教授及国家杰出青年杜勇、陈利、刘树红和李凯)、4名副教授(孔毅、汪炯、王建川、刘钰玲)、1名讲师(文诗艺)、2名博士后和60位研究生组成的科研队伍。黄伯云院士任研究室学术顾问。研究室同维也纳大学、瑞典皇家工学院、德国波鸿-鲁尔大学、美国宾州州立大学等国际20所著名大学建立了密切的科研合作关系。2003年以来主持国家自然科学基金创新研究群体、国家杰出青年科学基金、国家自然科学基金重点项目、国家863、973计划、重点研发计划、中德科学中心重大国际合作项目、企业开发项目等科研项目60项,科研经费总数近亿元。现每年面向国内外高校招收博士研究生8名左右,硕士研究生16名左右。
二、成员介绍

杜勇,教授,博导,相图、相变及合金设计

刘树红,研究员,博导,合金热力学、热物性质及应用

陈利,教授,博导,耐磨涂层

李凯,研究员,博导,铝合金微纳结构与力学性能

孔毅,副教授,博导,纳米结构及力学性能的多尺度计算

王建川,副教授,博导,储能材料的计算与设计

汪炯,副教授,博导,存储芯片材料的计算与设计

刘钰玲,副教授,博导,相图、扩散及微结构演变模拟

文诗艺,讲师,材料热物理性质及高通量计算方法、机器学习
三、研究方向和研究体系
研究方向:
1.材料智能设计软件+相图热力学(CALPHAD方法+实验测定)
2.材料智能设计软件+热物性质(扩散、界面能、热导率、体积、粘度)的实验测定和计算模拟
3.第一原理计算、分子动力学、Monte Carlo模拟
4.材料三维结构(结构模拟、有限元、TEM/3DAP等实验)
5.结构-性能相关性的理论和实验研究
6.集成计算材料工程(材料基因工程)
研究体系:
1.硬质合金、金属陶瓷和PVD, CVD耐摩涂层
2.铝基、镁基、铜基、钛基合金、贵金属、大宗工业固废(高值化)
3.能源材料(Li离子电池、太阳能材料、储氢材料)
4.核材料
四、代表性成果
4.1硬质合金、耐磨涂层研究:
(1)以WC-Ni硬质合金的关键子三元体系为研究对象,采用CALPHAD方法结合关键实验,开展了WC-Ni合金子体系的相图热力学和扩散动力学研究,进一步完善了硬质合金热动力学数据库CSUTDCC2,CSUDDCC2。基于数据库计算了一系列硬质合金的烧结碳窗口,并设计了合金成分和烧结工艺,探讨微量元素添加对WC-Ni硬质合金的微观结构及性能的影响,以制备耐腐蚀性能优异的WC-Ni硬质合金。
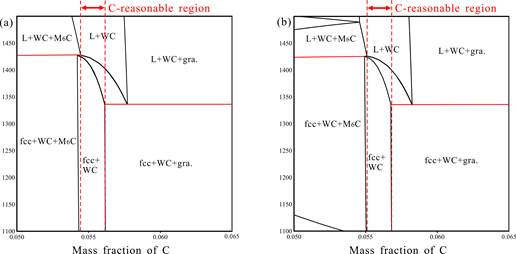
图1.热力学计算得到的W-C-Ni-Mo相图(a) WC-8.5%Ni-1.5%Mo合金的烧结碳窗口;(b) WC-8.0%Ni-2.0%Mo合金的烧结碳窗口
(2)围绕热导率预测模型的构建和计算软件的开发展开了一系列研究工作。基于微结构发展了描述两相复合材料热导率的模型,首次建立了WC的热导率随晶粒尺寸和温度变化的关系,并成功预测了WC/Co、WC/Ag和WC/WC界面的界面热阻,并成功应用于WC-Co-TiC三相硬质合金的热导率预测。该模型不仅考虑了温度、晶粒尺寸和相分数等因素,还首次引入了Co/(Ti,W)C界面热阻的概念,实现了对复杂多相系统热导率的精确预测。相关模型均已程序化并植入团队研发的CALTPP(CALculation of ThermoPhysical Properties)热物性计算软件中,相关研究人员可以利用该软件优化或获取目标材料体系的热导率数据。
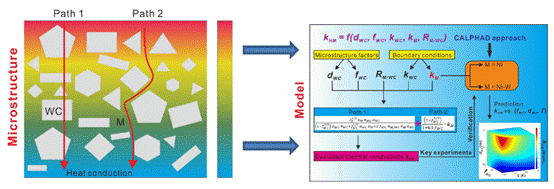
图2.基于微结构构建的两相复合材料热导率计算模型
(3)以化学气相沉积TiAlSiN涂层为研究对象,通过构建包含亚稳AlN相的Ti-Al-Si-N四元热力学数据库以及热力学计算,预测各沉积参数对涂层元素含量的影响。利用热力学计算指导沉积参数的选取,结合X射线衍射、透射电子显微镜和摩擦磨损试验等实验与表征手段,获得热稳定与抗高温氧化性能优异的新型CVD TiAlSiN涂层,为实际工业制备CVD涂层过程中工艺参数的选取与优化提供相关的理论基础与数据支撑。
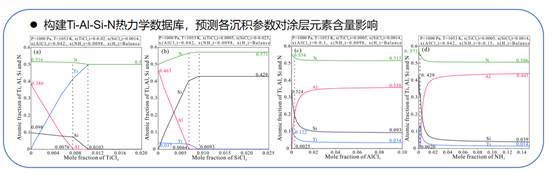
图3.热力学计算所得各沉积参数对CVD TiAlSiN涂层元素含量影响
(4)TiAlN涂层的多元合金化作为改善其切削性能的有效途径受到了广泛关注。基于关键实验结合第一性原理计算发现,在TiAlN涂层中添加少量氧抑制了Al扩散主导的调幅分解和随后的六方相变,从而显著提升了涂层的热稳定性能。相关成果发表在《Acta Materialia》,论文链接:https://doi.org/10.1016/j.actamat.2022.117706。
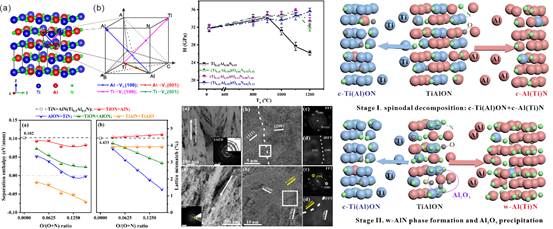
图4. TiAlON涂层的热分解行为和高温力学性能
(5)过渡族金属(TM)掺杂是提高TiAlN涂层性能的有效方式之一。本研究结合高通量第一性原理和机器学习建立了高效的TiAlTMN涂层成分设计策略,基于此开发了多种高性能TiAlTMN涂层,并在切削刀具上实现了应用。相关成果发表在《Acta Materialia》,论文链接:https://doi.org/10.1016/j.actamat.2024.120139。
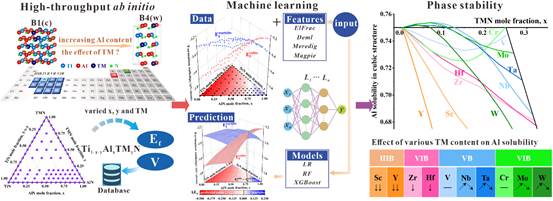
图5.基于高通量第一性原理和机器学习分析TiAlTMN涂层的立方相稳定性
4.2轻合金研究
(1)结合相图热力学计算,提出了一种耦合高通量析出模拟和关键实验的方法来优化合金成分和调控微观结构。在KWN(Kampmann-Wagner numerical)的框架下,系统研究了Al-Zn-Mg-Cu合金的成分/工艺-结构-性能关系,建立了一种新的材料设计策略。此外,基于相图热力学计算和合金元素协同强化概念,设计了Sc、Zr微合金化,不同Zn/Mg比的高镁铝合金。研究成分和工艺对高镁铝合金时效析出行为和力学性能的影响,为轻质高强铝合金的研发提供了参考。
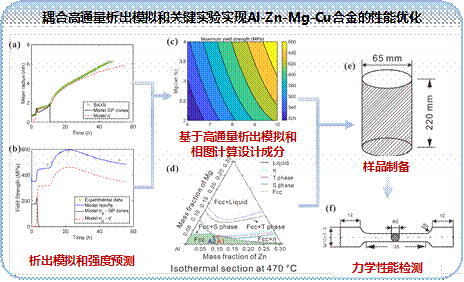
图6. Al-Zn-Mg-Cu合金成分设计和性能优化的研究思路
(2)结合透射电镜和第一性原理计算等实验和理论手段,重点深入研究铝合金纳米析出强化机理,并探索在该机理和相图计算(CALPHAD)指导下调控合金多尺度组织结构从而提升综合力学性能的方法。
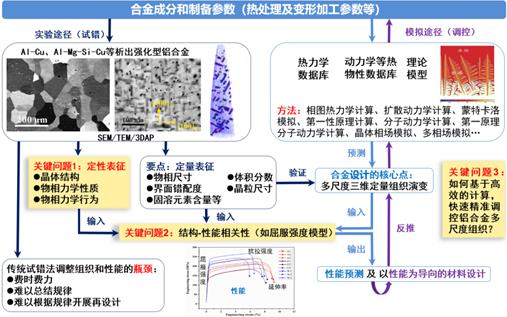
图7.近15年开展铝合金微米纳米尺度强化机理研究及多尺度组织调控的思路
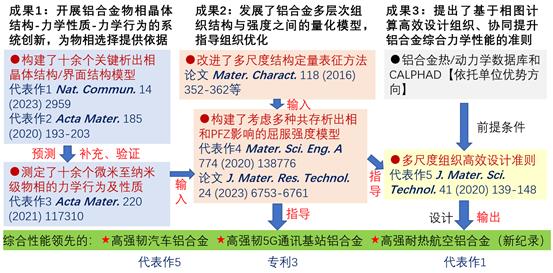
图8.“铝合金微纳结构表征与设计调控”研究方向围绕图1思路取得的三个方面的研究进展

图9.采用透射电镜观察结合第一性原理计算构建的结构模型举例。其中B′相的应力应变曲线为同行根据本组构建的结构模型模拟得到

图10.对铝合金纳米至微米尺度物相的力学行为/性质的研究成果举例。(a)Al-Mg-Si合金变形过程中β″被位错切过的TEM原位拉伸试验,(b)Al-Si-Mg合金变形过程中的SEM原位拉伸试验,(c)制备的毫米级超大第二相单晶颗粒及其(d)纳米压痕曲线和弹性模量测量值
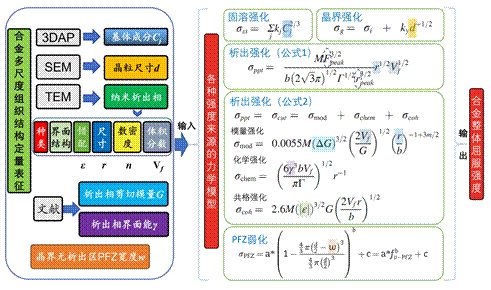
图11.基于多尺度组织结构定量表征开展的含多种析出相和PFZ影响下的铝合金强度预测。组织定量表征为强度预测提供了可靠的输入参数
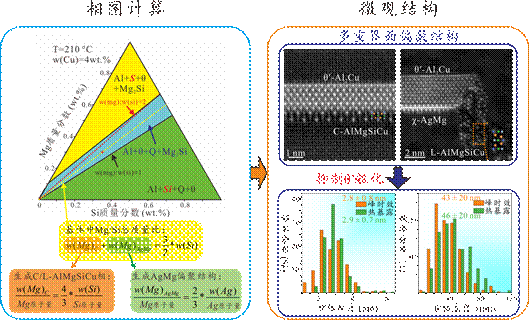
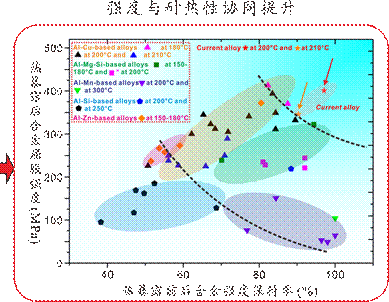
图12.研究组进行耐热高强铝合金设计的案例示意图。基于相图计算来分步调控成分,在避免高温易粗化S相以及硬脆Si相的生成的同时优化Mg/Si元素含量比,再根据已选定的Ag和Mg/Si比例,确定Mg和Si的具体添加量,促进含有C/L相以及AgMg偏聚结构的多重界面结构生成,实现对主强化相θ′的有效包覆,极大地抑制了析出相的粗化,而在时效热处理过程早期形核的C/L相可以作为中期形核的θ′相的异质形核点,提高了θ′的数密度,因而合金实现了强度与耐热性的协同提升[Nat. Commun. 14 (2023)2959]
(3)基于平衡合金法及物相表征实验及相图计算方法研究了Mg-Zn、Zn-Mn、Mg-Zn-Mn和Mg-Al-Zn体系的热力学参数并构建了Mg-Al-Zn-Mn四元体系的热力学数据库。通过热力学计算与腐蚀形貌观察、失重测试和电化学测试相结合研究Zn、Mn单独添加及Al、Zn、Mn任意两种元素同时添加对Mg合金腐蚀行为的影响。通过相图计算方法计算了多种成分Mg-Al-Zn-Mn合金的凝固路径,通过分析不同元素在(Mg)基体中的固溶度、中间相的种类和含量、中间相的相成分以及中间相与基体间的电位差,从热力学角度设计了发生电偶腐蚀倾向最小的Mg-Al-Zn-Mn合金,并根据计算结果选取了另外两个合金成分作为对比。在此基础上,通过微观组织分析和物相表征证明了所建立数据库的准确性。通过腐蚀形貌观察、失重测试以及电化学测试分析了各合金的腐蚀行为,验证了热力学计算在Mg合金腐蚀行为研究及合金成分设计中的可行性。
图13. Mg-Al-Zn-Mn合金热力学及腐蚀行为研究:(a)基于数据库计算的Mg-0.095Al-0.0084Zn-Mn垂直截面与实验数据的比较;(b)计算的Mg-Al-Zn-Mn合金中Mg17Al12相和LTAl8Mn5相的摩尔分数;(c)基于数据库设计的Mg-Al-Zn-Mn合金在3.5 wt.% NaCl溶液中的动电位极化曲线.
(4)弹性模量通常用来评价材料的力学性能,如塑性、脆性、强度、硬度等。如何有效并可靠的计算两相及三相以上合金的弹性模量,对于合金材料的设计研发具有重要意义。本工作基于第一性原理计算和CALPHAD方法提出了一种计算有限温度下多相合金弹性模量的方法。首先,基于两种极限提出了多相合金弹性模量的计算模型;其次,考虑温度和固溶度通过第一性原理计算单相的刚度矩阵;然后,通过CALPHAD方法计算合金的相分数和温度,获得多相合金在有限温度的刚度矩阵,从而实现多相合金弹性模量的高效计算。计算方法的可靠性通过实验测得的两相和三相Mg-Al-Si合金的弹性模量得以验证。基于该计算方法,我们对Mg-Al-Si合金623K宽广成分的弹性模量进行了深入描述,并预测了高弹性模量的成分范围。
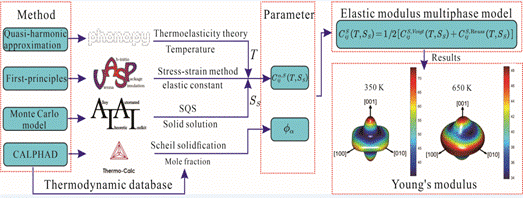
图14.多元多相合金弹性模量的计算和实验验证
4.3铜基、大宗工业固废(高值化)
(1)在优化包含有序无序转变的Cu-Be体系时,基于密度泛函理论计算了有序相bcc_B2的形成焓,并提出了分步的热力学优化步骤。提出的分步优化方法可望为其他包含有序无序转变的复杂体系热力学优化提供指导。此外,基于CALPHAD方法,构建了包含26个元素的铜合金体系fcc相和液相的扩散动力学数据库。结合计算模拟和实验研究开展了Cu-Cr系合金的微结构和性能研究。
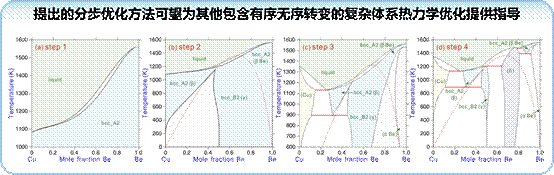
图15. Cu-Be体系分步的热力学优化步骤
(2)以大宗铝硅酸盐固废中典型氧化物Al2O3、CaO、FeOX、MgO、Na2O和SiO2为研究对象,采用CALPHAD方法和关键相图实验研究,开展了Al2O3-CaO-FeOX-MgO-Na2O-SiO2体系相关二元系和三元系的相图热力学研究。在子体系的相图热力学基础上,构建了六元铝硅酸盐Al2O3-CaO-FeOX-MgO-Na2O-SiO2相图热力学数据库,实现相图、热力学性质、相变驱动力和凝固过程等的计算模拟,为固废材料化应用提供指导。
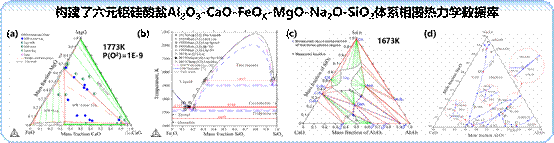
图16.氧化物体系的相图和热力学研究
4.4能源材料及储能材料
(1)以锡基负极为研究对象,采用CALPHAD方法、第一性原理计算和实验研究的结果参数,开展了
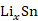 理想球型负极颗粒的充放电循环过程的模拟研究。在计算过程中,考虑了Li-Sn体系中的纯
理想球型负极颗粒的充放电循环过程的模拟研究。在计算过程中,考虑了Li-Sn体系中的纯
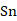 、
、
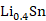 、
、
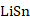 、
、
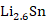 、
、
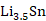 五个相之间的转变,模拟了一个完整的充放电单循环。将充放电速率、相分布、弹性以及电压曲线联系起来,解释电压曲线表现的原因,为电极材料设计提供指导。
五个相之间的转变,模拟了一个完整的充放电单循环。将充放电速率、相分布、弹性以及电压曲线联系起来,解释电压曲线表现的原因,为电极材料设计提供指导。
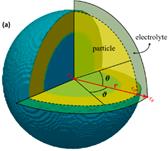
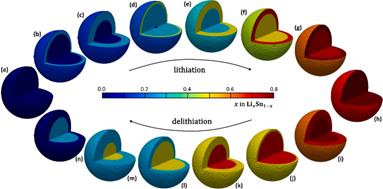
图17.
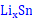 理想球型负极颗粒的充放电循环过程的建模(左图)及模拟研究(右图)
理想球型负极颗粒的充放电循环过程的建模(左图)及模拟研究(右图)
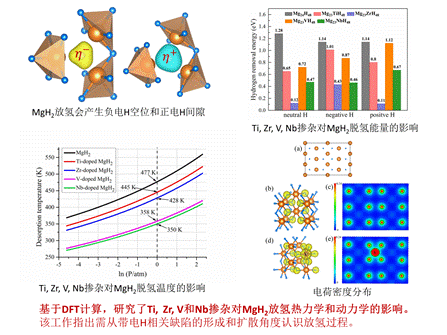
(2)认识MgH2的脱氢热力学和动力学对其工程应用至关重要,掺杂是改善MgH2放氢热力学和动力学性能的常用方法。利用第一原理计算,我们从点缺陷角度研究了Ti、Zr、V和Nb掺杂对MgH2放氢性质的影响。研究发现,MgH2放氢过程中出现的氢空位和氢间隙会以带电状态存在,而这四种掺杂剂更倾向于以中性状态替代Mg,纯的MgH2中氢的传输以带正电的氢空位或带负电的间隙氢为主;电子结构分析表明,这四种掺杂剂可以弱化较强的Mg-H离子键;热力学性质计算表明Ti、Zr、V和Nb掺杂不仅能降低其附近中性和带电H原子的微观脱氢能,还能降低MgH2的宏观脱氢温度;在掺杂MgH2中,带电氢间隙很难从掺杂原子周围逃逸,但正电氢空位和负电氢空位却很容易从掺杂原子周围逃逸,表明掺杂原子在MgH2脱氢过程中不会阻碍氢传输。该研究的主要强调从点缺陷研究MgH2脱氢性能时需考虑缺陷带电。
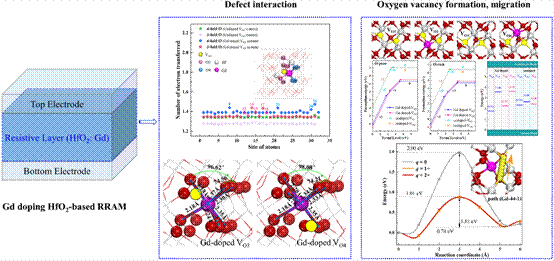
(3)HfO2掺杂Gd调控氧空位细丝的导电机理及器件性能优化研究,Gd掺杂可以有效调控氧空位的随机分布以及导电细丝的随机形成和断裂,提高器件的参数均一性能。Gd掺杂不仅会显著降低其周围氧空位的形成能,即氧空位在能量上更倾向于在Gd原子周围形成;还会使其周围带+2q电荷的氧空位的迁移能垒明显增加,从而在能量上限制氧空位的随机迁移,抑制氧空位导电细丝的随机形成和断裂。同时掺杂剂Gd周围原子之间的电荷转移大大增强,库仑相互作用更加显著,器件的导电性能将明显提高
4.5工业软件
(1)外加磁场下的相图相变研究:为研发磁场下具有应用潜力的新型材料,从热力学和材料设计角度深入开展科学研究工作,在磁场下热力学模型、相图热力学计算和微观结构模拟等方面取得了系列创新成果。集成Weiss分子场理论、数值计算和相图计算方法,只基于材料的磁性参数,建立了外加磁场下适用于纯组元和化学计量比化合物的通用热力学模型。基于建立的磁场热力学模型,定量计算磁场对Bi-Mn二元系相图热力学的影响,根据理论计算提出制备大量高纯度BiMn切实可行的方法。针对定量模拟外加磁场下合金热处理过程中的微观结构演变这一严峻挑战,构建耦合真实热力学能量的相场模型,实现定量模拟Fe-C合金在热磁处理过程中形成的取向结构。
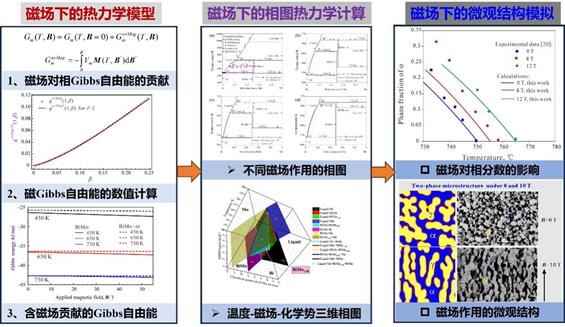
图18.外加磁场下热力学模型、相图热力学计算和微观结构模拟研究
(2)热物性研究:推导出了一个通用的热力学模型用于计算液/液和固/固共格界面能。George Kaptay教授在其新发表的论文致谢中提到“特别感谢刘钰玲女士指出作者以前一些关于界面能论文的矛盾之处(Adv. Colloid. Interface Sci., 2020, 283: 102212)”。开发了一款计算和优化热物性参数的软件CALTPP (CALculation of ThermoPhysical Properties),可以计算随成分和温度变化的扩散系数、体积、界面能、热导率和粘度,为结构模拟提供关键输入参数。

图19.热物理性质计算软件CALTPP的框架图
(3)晶体塑性有限元研究:以6XXX系列铝合金中关键析出相对力学性能影响关系为研究对象,采用晶体塑性理论和多尺度计算方法,进行了6XXX铝合金的力学性能多尺度模拟研究。在晶体塑性理论的基础上,构建了从微观组织演变到宏观力学响应的全序列计算模型,实现了析出相尺寸分布、体积分数、应力-应变曲线和加工硬化行为的模拟预测,为高性能铝合金的设计和工业生产提供了理论支撑和计算工具。
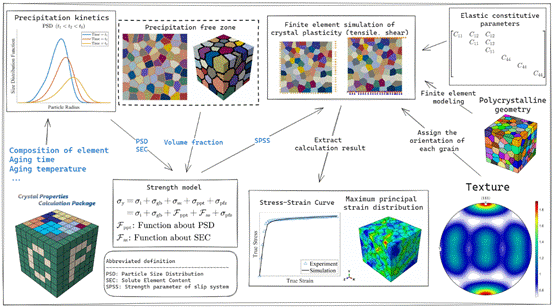
图20. 6XXX时效强化铝合金微观结构-力学性能多尺度模拟图


